
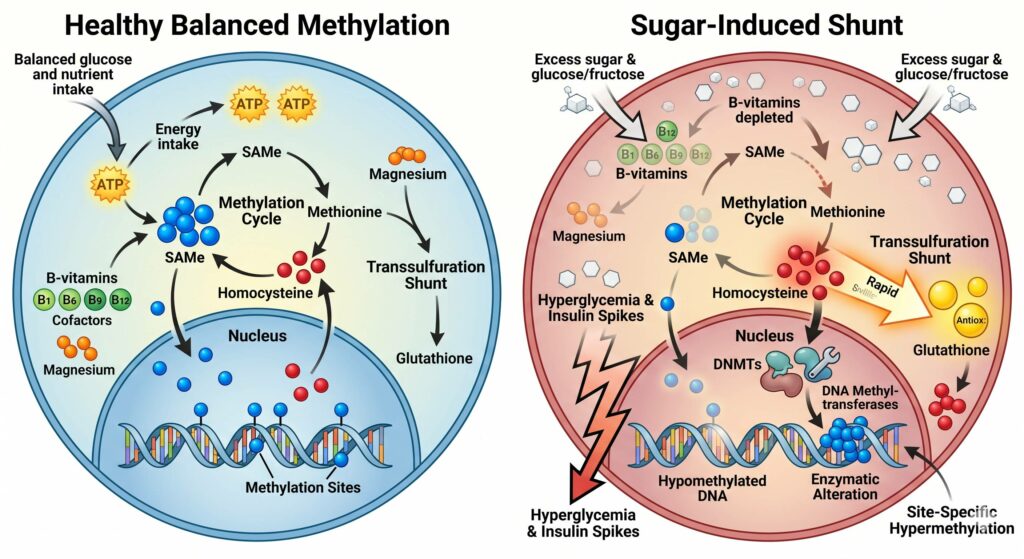
Chronic consumption of refined sugar exerts a significant “biochemical tax” on the body’s methylation capacity. This occurs not through a single mechanism, but through a cascade of cofactor depletion, oxidative stress, and altered enzymatic signaling.
1. Cofactor Depletion and Competition
The metabolism of glucose requires several of the same micronutrients that drive the one-carbon cycle. When sugar intake is chronically high, the body prioritizes glycolysis and the citric acid cycle to process energy, leading to a functional deficiency in key methylation cofactors:
- B-Vitamins: Processing high sugar loads rapidly consumes B1, B2, B3, and B6. B6 (Pyridoxal 5′-phosphate) is particularly critical because it acts as a cofactor for Cystathionine beta-synthase (CBS), the enzyme that initiates the transsulfuration pathway.
- Magnesium: Required for the synthesis of S-Adenosylmethionine (SAMe), the body’s universal methyl donor. Sugar consumption increases renal magnesium excretion, potentially limiting the conversion of Methionine to SAMe via the enzyme methionine adenosyltransferase (MAT).
2. The Oxidative Stress Pivot (Transsulfuration)
Chronic sugar intake leads to the formation of Advanced Glycation End-products (AGEs) and increased reactive oxygen species (ROS). This creates a state of oxidative stress that fundamentally shifts the direction of the methylation cycle:
- The Glutathione Demand: When cells are under oxidative stress, the body prioritizes the production of Glutathione, the master antioxidant.
- Homocysteine Diversion: Instead of being remethylated back into Methionine (which requires B12 and Folate), Homocysteine is “shunted” down the transsulfuration pathway to create Cysteine and eventually Glutathione.
- Result: While this helps combat oxidative stress, it reduces the availability of Methionine for the regeneration of SAMe, effectively “thinning” the methyl donor pool available for DNA methylation and neurotransmitter synthesis.
3. Impact on DNA Methyltransferases (DNMTs)
Hyperglycemia and the resulting hyperinsulinemia can directly alter epigenetic programming. High circulating insulin levels have been shown to influence the expression and activity of DNA Methyltransferases (DNMTs), the enzymes responsible for placing methyl groups on DNA.
Chronic sugar consumption is often associated with:
- Global Hypomethylation: A general decrease in methylation across the genome, which can lead to genomic instability.
- Site-Specific Hypermethylation: The “silencing” of specific promoter regions, particularly those involved in metabolic regulation and anti-inflammatory responses.
4. The Role of Uric Acid
High intake of fructose (a component of sucrose and high-fructose corn syrup) raises systemic uric acid levels. Elevated uric acid is known to induce mitochondrial oxidative stress and inhibit the activity of certain enzymes in the one-carbon cycle, further compounding the inhibition of proper methylation.
Summary of the Biochemical Shift
The relationship can be visualized as a shift in the following equilibrium:
Homocysteine –> 5-MTHF –> B12 –> Methionine –> SAMe –> Methylation
Under high-sugar conditions, the pathway is forced toward:
Homocysteine –> B6/Oxidative Stress –> Cystathionine –> Cysteine –> Glutathione
This depletion of the methyl pool can lead to long-term changes in gene expression, impaired detoxification, and reduced neurotransmitter turnover.
Chronic sugar consumption imposes a substantial “biochemical tax” on the body’s methylation capacity by depleting essential cofactors and shifting metabolic priorities. To process high glucose loads, the body rapidly consumes B-vitamins and magnesium, diverting these nutrients away from the one-carbon cycle where they are needed to synthesize S-Adenosylmethionine (SAMe), the universal methyl donor. This nutrient drain is compounded by sugar-induced oxidative stress, which triggers a “transsulfuration shunt.” Instead of recycling homocysteine into methionine to support methylation, the body prioritizes the production of the antioxidant glutathione, effectively thinning the available pool of methyl groups for DNA regulation and neurotransmitter synthesis.
Beyond nutrient depletion, chronic hyperglycemia and hyperinsulinemia directly disrupt the enzymatic machinery of epigenetics. Elevated insulin levels can alter the activity of DNA Methyltransferases (DNMTs), leading to a state of global hypomethylation that threatens genomic stability while simultaneously “silencing” specific genes through site-specific hypermethylation. Furthermore, the metabolic byproduct of fructose – uric acid – induces mitochondrial stress that further inhibits the one-carbon cycle. Collectively, these mechanisms illustrate how a high-sugar diet can fundamentally rewire the body’s epigenetic landscape, impairing detoxification and metabolic efficiency over time.
Leave a Reply